

Fribourg Lab Research
Multidisciplinary approaches to understand, monitor, and modulate T cell function




Research Mission & Focus
T cells are critical for fighting infection and cancer and maintaining immune tolerance. Effector T cells (Teff) directly defend us from pathogens and eradicate cancerous cells. Regulatory T cells (Treg) shut down the immune response when the threat is gone and prevent deleterious immune responses against self. Disruption of the Treg to Teff balance underpins the pathology of autoimmune disease, is exploited by tumors in malignancy, and represents a barrier in transplantation.
Donor-reactive Teff cause transplant rejection and are targeted by most immunosuppressive drugs currently administered to transplant recipients in the clinic (e.g., calcineurin inhibitors, mycophenolate mofetil, steroids). Regulatory T cells (Treg) are also critical in controlling these donor-reactive T cell responses in organ transplant recipients. While a balanced ratio of regulatory T cells to donor-reactive effector T cells (Treg/Teff) is essential to prevent allograft rejection, accurately determining this balance in each individual transplant recipient remains an unmet challenge. This is because the activity and function of both Teff and Treg (reactivity for Teff, and suppressive capacity for Treg) is regulated by multiple dynamical processes and varies significantly across individuals and within cells from the same T cell compartment.
The Fribourg Lab research program aims to unravel the mechanisms that govern this delicate Treg/Teff balance and leverage this knowledge to develop innovative approaches to monitor and modulate T cell function and ultimately guide therapeutic intervention.
Research Directions
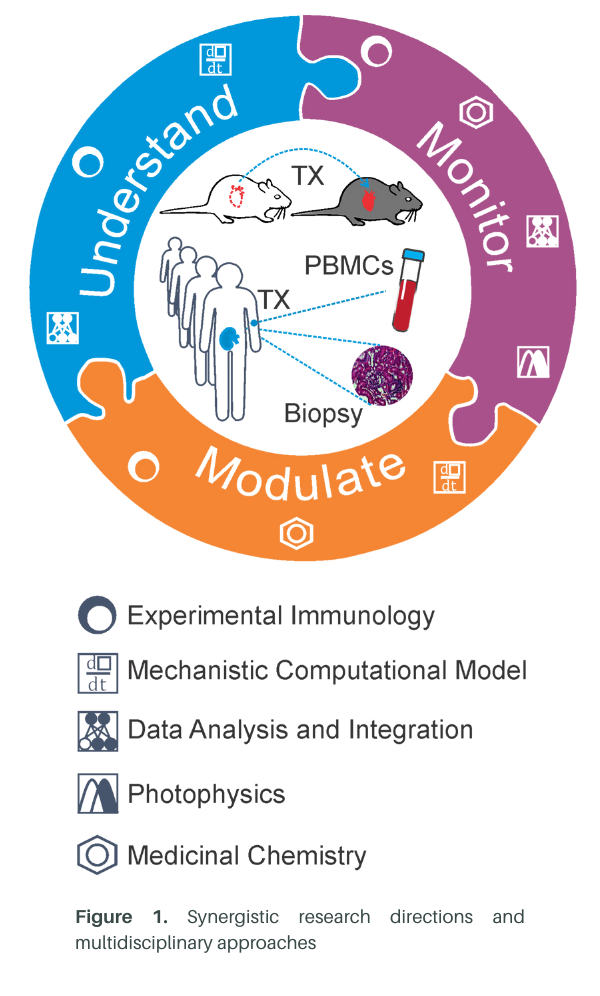
Since establishing the Fribourg Lab research group in 2018, fostering innovation has been one of its core tenets. We have ensured that all members have cross-disciplinary training in addition to immunology (e.g., medicinal chemistry, pharmacology, engineering) and created a hub of scientists that, together with clinicians, aim to solve problems in transplantation and immunology at large coming from different perspectives and with unique ideas. Our research program is structured around three synergistic pillars or research directions (Fig. 1)
A. Understanding the mechanisms that govern the Treg/Teff balance.
Challenge: T cells receive a multitude of signals (e.g., T cell receptor activation, cytokines, metabolic cues) that need to be integrated at the cellular level to decide on fate and function. Our efforts are concentrated on studying this signal integration in systems engineering terms to identify molecular mechanisms that have differential effects on Treg and Teff cells.
Multidisciplinary approach: We are using a combination of mouse transplant models, in vitro assays of murine and human T cell function, as well as ordinary differential equation (ODE) and agent-based (AB) computational mechanistic models of T cell polarization to identify novel pathways that modulate T cell function.
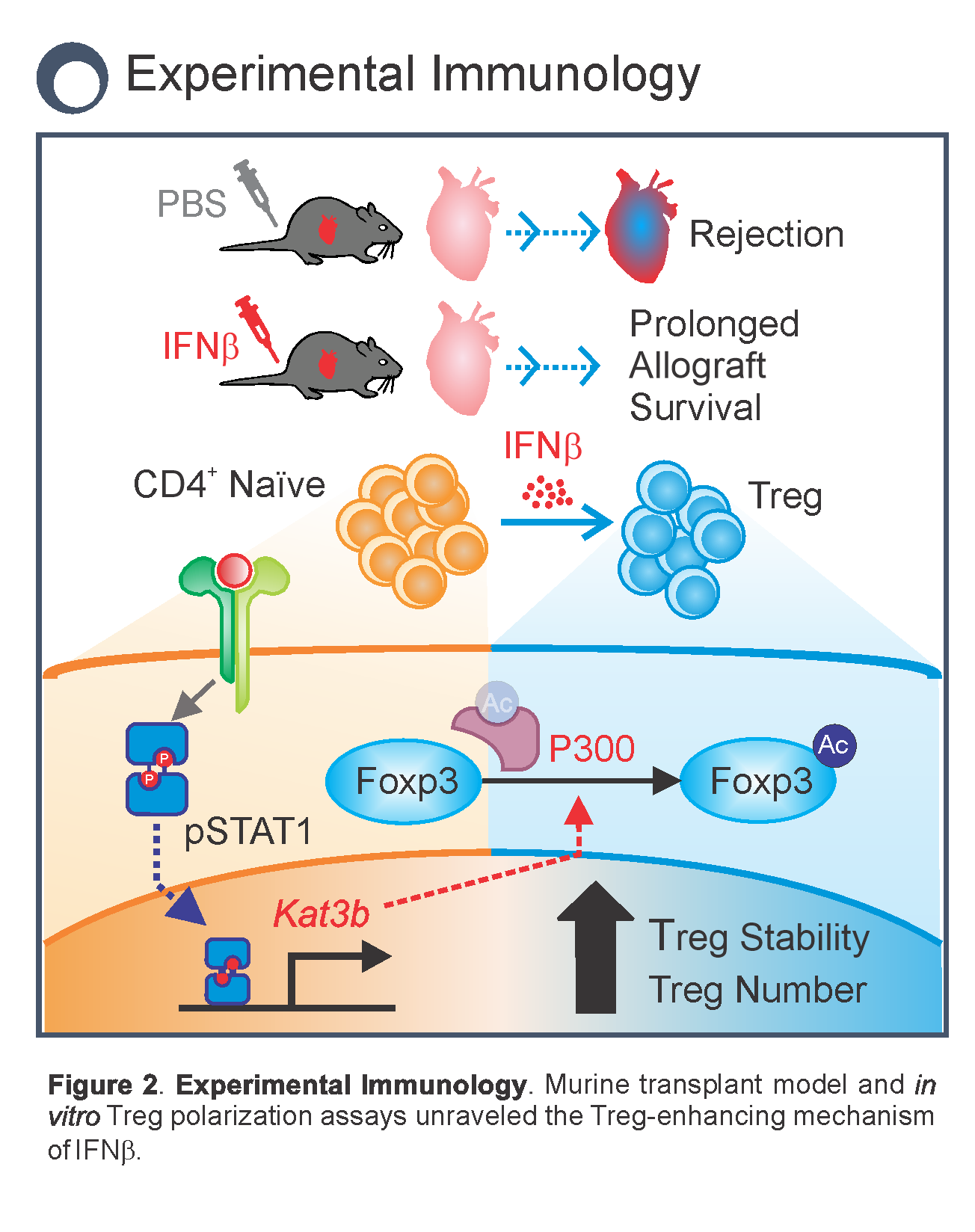
Impact: Type I interferons (IFN) are a family of pleiotropic antiviral/inflammatory cytokines, whose role in innate immunity has been widely characterized. Strikingly, their effects on adaptive immunity appear to switch from pro-inflammatory to immunosuppressive depending on context. Interferon-beta (IFNβ) has been used to treat remitting-relapsing sclerosis patients since the 90s, but its immunosuppressive mechanism remained elusive. Using our multidisciplinary approach, we have uncovered a mechanism through which IFNβ enhances Treg activity and established its potential application in transplantation. Simulations of the effects of IFNβ on T cell polarization in silico yielded the paradigm-shifting prediction that direct effects of IFNβ on T cells could promote Treg. The computational model, containing 94 species and 53 chemical reactions, also provided a molecular mechanistic hypothesis whereby IFNβ promotes acetylation of Foxp3, the master transcription factor regulator in Treg, to stabilize it and increase its suppressive function. We tested and confirmed this mechanism first in Treg induction cultures and then in a Treg-dependent mouse transplant model, and established the ability of exogenous IFNβ to prolong allograft survival. (Fig. 2 and Fig. 3)
We also investigated the interplay between IFNβ and interleukin-6 (IL-6), a key inflammatory cytokine in autoimmune disease, and ischemia-reperfusion injury known to hinder Treg induction. Mechanistically, we demonstrated that IFNβ and IL-6 signal independently to promote opposing effects on the acetylation of Foxp3. We further showed that this mechanism is conserved in both murine and human cells, underscoring the importance of this finding in immune regulation (manuscript under review).
IFNs signal through the JAK/STAT-coupled type I interferon receptor formed by two transmembrane subunits, IFNAR1 and IFNAR2 (classic signaling). We have recently identified a previously unrecognized mode of IFN signaling (trans-signaling) that uses IFNAR1 but involves a soluble isoform of the interferon receptor 2 subunit (sIFNAR2) and obtained data indicating its potent effects in shifting T cell responses towards regulation in murine in vivo models in two different contexts (adoptive T cell transfer colitis and heart transplant) supporting the broad relevance of this mechanism (manuscript under preparation).
Our findings have unveiled unrecognized links between innate and adaptive immunology. They have also spurred a strong interest in understanding the dynamics of the acetylome, the complete set of acetylated proteins in the cell, and its impact on T cell polarization. This has driven numerous subsequent projects in the lab focused on both monitoring (B) and modulating acetylation of key proteins in T cells (C).
Funding: R01-AI14710, R56-AI141710.
B. Monitoring T cell function in clinical samples.
Challenge: High-dimensional single-cell techniques have revealed a remarkable heterogeneity within the T cell compartment. The challenge lies in linking this heterogeneity to T cell function and developing user-friendly assays for clinical settings, to allow monitoring of T cell function status in patients. This is crucial for transplant recipients, who walk a tightrope between preventing graft rejection and managing immunosuppression-related complications like malignancy and infection.
Multidisciplinary approach: We use high-dimensional omics tools and data integration computational analyses to identify key phenotypic, transcriptional, and T cell receptor (TCR) repertoire features in peripheral blood T cells and biopsies from kidney transplant recipients. We pair these analyses with chemical synthesis and photophysics to develop novel fluorescent biosensors to assay T cell function compatible with flow cytometry.
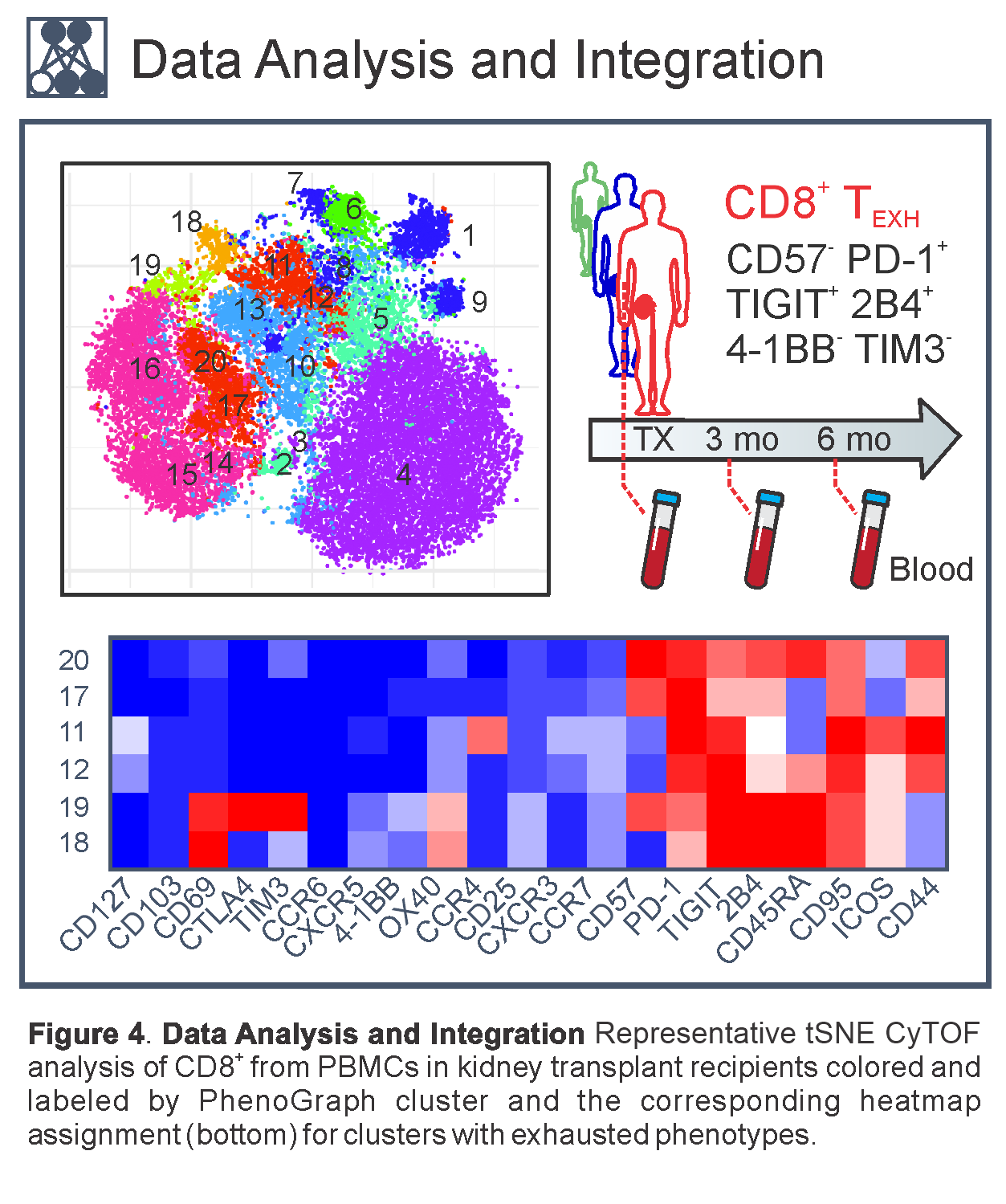
Impact: We have been performing mechanistic studies with samples from the NIAID Clinical Trials in Organ Transplantation (CTOT) consortium since 2018. We identified a pro-apoptotic T cell transcriptomic signature associated with successful withdrawal of the calcineurin inhibitor tacrolimus.6 We characterized PD-1+ CD57- TIGIT+ 4-1BB- TIM3- exhausted T cells as a potential biomarker for improved allograft function following lymphocyte-depleting induction therapy with rabbit anti-thymoglobulin (rATG)7 (Fig. 4). We demonstrated that while rATG effectively depletes CD4+ T cells, a subset of donor-reactive CD8+ T cells can escape depletion and potentially mediate graft damage. We also developed pipelines and analyzed spatial transcriptomics in kidney biopsies and extracted focal features of acute T cell mediated rejection.
We participated in the mechanistic studies of COVID-19 Protection After Transplant (CPAT) trials funded by the National Institutes of Health to investigate the safety and immunogenicity of SARS-CoV-2 vaccination strategies in solid organ transplant recipients. In these individuals, we observed significantly reduced spike-specific CD8+T cell receptor repertoire depth in kidney transplant recipients compared to healthy controls following vaccination, despite comparable CD4+ spike-specific T cell responses.
Inspired by our Treg mechanistic studies, we have developed an assay to measure protein acetylation compatible with flow cytometry. We have used this FRET-based method to obtain information per cell regarding the extent of Foxp3 acetylation (associated with Treg suppressive function), selectively in the Foxp3+ compartment.
Treg rely on a unique metabolic balance and signature for their suppressive function. Cellular reduction-oxidation (redox) homeostasis mechanisms are fully intertwined with the metabolic state, and thus this metabolic signature results in a specific ratio of glutathione (GSH) and its precursor, gamma-glutamylcysteine (γ-GC). Current methods for assessing this balance in live T cells are inadequate. Existing assays measure γ-GC in cell lysates, lack specificity, or irreversibly bind GSH, disrupting cellular homeostasis and masking heterogeneity. To address these limitations, we first developed FJF375, a novel fluorescent probe for real-time, reversible GSH detection in live cells. Our preliminary data demonstrate its suitability for assessing single-cell redox homeostasis in PBMCs with minimal impact on cellular function. We are currently conducting pilot studies in kidney transplant recipient clinical samples from previous studies at different clinical centers (see collaborations below) to standardize the assay and establish power analysis calculations for future prospective studies. In addition, we have designed and are now synthesizing a dual γ-GC/GSH (Fig. 5) sensor to investigate the hypothesis that variations in GSH homeostasis within Treg population drive functional heterogeneity. This approach holds significant promise for understanding T cell function at large, and Treg function in particular, in clinical settings.
Funding: U01-AI388897, R21-AI83028
C. Modifying T cell function through novel pharmacological approaches.
Challenge: As part of our mechanistic findings and transcriptomic analyses in clinical samples, we have identified post-translational modifications in Foxp3 (acetylation), p53 (acetylation), and AMPK substrates (phosphorylation) that are key for T cell polarization and function. The current challenge is to be able to selectively modulate protein acetylation at these sites in T cells therapeutically.
Multidisciplinary approach: We are using chemical design and synthesis together with computational modeling (structural and mechanistic) to design new small molecules to promote targeted protein post-translational modifications in T cells and testing their modulatory properties in murine and human in vitro T cell assays, and in vivo murine models.
Impact: Foxp3 acetylation is mediated by two acetyltransferases in the cell, P300 and TIP60. We identified that TIP60 inhibitors, contrary to expectation, augment Foxp3 acetylation and Treg cell induction. Using a combined computational and experimental approach, we discovered that low-dose TIP60 inhibitors paradoxically activate TIP60 to acetylate P300, which in turn, increases Foxp3 acetylation, promoting Treg cell generation. (Fig. 6)
The three pillars of the Fribourg Lab research program (Fig. 1) perfectly align with our medical mission to identify the molecular drivers of immune imbalance behind human disease. Led by Miguel Fribourg, PhD, the research program benefits from the synergy between disciplines and clinicians, which requires listening with the understanding that depth of knowledge takes time and dedication and that there is value in approaching a problem from different perspectives. This guides our focus on mentoring talented researchers from diverse training backgrounds, countries, and cultures, to instill multidisciplinarity as a core competency for future immunologists.




Fribourg Lab PUBLICATIONS
